الثلاسيميا
Thalassemia
تلاسيميا
الثلاسيميا هو اضطراب وراثي في الدم أي ينتقل من الآباء إلى الأطفال من خلال الجينات ويحدث عندما لا ينتج الجسم ما يكفي من بروتين يسمى الهيموغلوبين وهو جزء مهم من خلايا الدم الحمراء، عندما لا يكون هناك ما يكفي من الهيموجلوبين لا تعمل خلايا الدم الحمراء في الجسم بشكل صحيح وتستمر لفترات زمنية أقصر؛ لذلك هناك عدد أقل من خلايا الدم الحمراء السليمة التي تنتقل في مجرى الدم.
تنقل خلايا الدم الحمراء الأكسجين إلى جميع خلايا الجسم، الأكسجين هو الوقود الذي تستخدمه الخلايا لتعمل وعندما لا يكون هناك ما يكفي من خلايا الدم الحمراء السليمة، لا يتم أيضًا توصيل كمية كافية من الأكسجين لجميع خلايا الجسم الأخرى، مما قد يتسبب في شعور الشخص بالتعب، أو الضعف، أو ضيق التنفس.
هذه حالة تسمى فقر الدم حيث قد يعاني الأشخاص المصابون بالثلاسيميا من فقر دم خفيف أو شديد، ويمكن لفقر الدم الشديد أن يتلف الأعضاء ويؤدي إلى الوفاة.
إن الحامل لمرض الثلاسيميا يكتسب مناعة ضد مرض الملاريا، واحتمالات النجاة من الملاريا ترتفع في المناطق المعروفة بانتشار مرض الملاريا؛ ولهذا السبب فإن هذا المرض منتشر في المناطق الإستوائية والمناطق القريبة منها.
أعراض الثلاسيميا
هناك عدة أنواع من مرض الثلاسيميا حيث تعتمد العلامات والأعراض التي تعاني منها على نوع حالتك وشدتها، ويمكن أن تشمل علامات وأعراض الثلاسيميا ما يأتي:
- تعب.
- ضعف.
- جلد شاحب أو مصفر.
- تشوهات عظام الوجه.
- النمو البطيء.
- انتفاخ البطن.
- البول الداكن.
تظهر على بعض الأطفال علامات وأعراض مرض الثلاسيميا عند الولادة حيث يقوم الآخرون بتطويرها خلال العامين الأولين من الحياة، وبعض الأشخاص الذين لديهم جين هيموغلوبين واحد مصاب لا تظهر عليهم أعراض مرض الثلاسيميا.
أسباب وعوامل خطر الثلاسيميا
تشمل أبرز أسباب وعوامل خطر الإصابة بالثلاسيميا ما يأتي:
1. أسباب الإصابة بالثلاسيميا
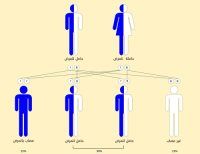
ينتج مرض الثلاسيميا عن خلل في الجينات التي تؤثر على إنتاج الهيموغلوبين، حيث لا يمكن أن يولد الطفل مصابًا بالثلاسيميا إلا إذا ورثوا هذه الجينات المعيبة من كلا الوالدين.
على سبيل المثال، إذا كان لدى كلا الوالدين الجين المعيب الذي يسبب ثلاسيميا بيتا الكبرى، فهناك احتمال واحد من كل 4 أن يولد كل طفل بهذه الحالة، حيث عادةً يكون والدا الطفل المصاب بالثلاسيميا حاملين للثلاسيميا، وهذا يعني أن لديهم واحدًا فقط من الجينات المعيبة.
2. عوامل خطر الإصابة بالثلاسيميا
تتضمن العوامل التي تزيد من خطر الإصابة بالثلاسيميا ما يأتي:
- تاريخ عائلي للإصابة بالثلاسيميا: ينتقل مرض الثلاسيميا من الآباء إلى الأطفال من خلال جينات الهيموغلوبين الطافرة.
- العرق: يحدث مرض الثلاسيميا في أغلب الأحيان عند الأمريكيين من أصل أفريقي وفي الأشخاص المنحدرين من منطقة البحر الأبيض المتوسط وجنوب شرق آسيا.
مضاعفات الثلاسيميا
تشمل المضاعفات ما يأتي:
1. المضاعفات المتوسطة للثلاسيميا
تشمل المضاعفات المحتملة لمرض الثلاسيميا المتوسطة إلى الشديدة ما يأتي:
-
الحديد الزائد
يمكن للأشخاص المصابين بالثلاسيميا الحصول على الكثير من الحديد في أجسامهم إما من المرض أو من عمليات نقل الدم المتكررة، لذا يمكن أن يؤدي الإفراط في تناول الحديد إلى تلف القلب، والكبد، ونظام الغدد الصماء والذي يتضمن الغدد المنتجة للهرمونات التي تنظم العمليات في جميع أنحاء الجسم.
-
العدوى
يعاني الأشخاص المصابون بالثلاسيميا من زيادة خطر الإصابة بالعدوى وهذا صحيح بشكل خاص إذا كنت قد خضعت لعملية استئصال الطحال.
2. المضاعفات الشديدة للثلاسيميا
في حالات الثلاسيميا الشديدة، يمكن أن تحدث المضاعفات الآتية:
-
تشوهات العظام
يمكن أن يؤدي مرض الثلاسيميا إلى تمدد نخاع العظام، مما يؤدي إلى اتساع عظامك حيث يمكن أن يؤدي هذا إلى بنية عظام غير طبيعية وخاصةً في وجهك وجمجمتك، كما قد يؤدي توسع النخاع العظمي أيضًا إلى جعل العظام رقيقة وهشة؛ مما يزيد من فرصة كسر العظام.
-
تضخم الطحال
يساعد الطحال جسمك على محاربة العدوى وتصفية المواد غير المرغوب فيها، مثل: خلايا الدم القديمة أو التالفة حيث غالبًا يصاحب مرض الثلاسيميا تدمير عدد كبير من خلايا الدم الحمراء، ويؤدي هذا إلى تضخم الطحال والعمل بجهد أكبر من المعتاد.
يمكن أن يؤدي تضخم الطحال إلى تفاقم فقر الدم، ويمكن أن يقلل من عمر خلايا الدم الحمراء المنقولة، لذا في حال نمو الطحال بشكل كبير جدًا فقد يقترح طبيبك إجراء عملية جراحية لإزالته.
-
تباطؤ معدلات النمو
يمكن أن يؤدي فقر الدم إلى إبطاء نمو الطفل وتأخير البلوغ.
-
مشاكل قلبية
يمكن أن يترافق قصور القلب الاحتقاني واضطراب نظم القلب مع الثلاسيميا الشديدة.
تشخيص الثلاسيميا
في معظم الحالات، يتم تشخيص ألفا ثلاسيميا من خلال فحص حديثي الولادة وهو فحص دم يُعطى عند ولادة الطفل لأول مرة، حيث قد يُعاني الأطفال المصابون بالثلاسيميا الكبرى من انتفاخ في البطن، أو أعراض فقر الدم، أو فشل النمو.
إذا اشتبه الطبيب في ثلاسيميا ألفا فسيأخذ عينة دم للاختبار حيث يمكن أن تكشف اختبارات الدم عن وجود خلايا دم حمراء شاحبة ومتنوعة في الشكل والحجم أو أصغر من الطبيعي، يمكنهم أيضًا اكتشاف انخفاض عدد خلايا الدم الحمراء والخلايا ذات التوزيع غير المتكافئ للهيموغلوبين، مما يجعلها تبدو مثل عين الثور عند رؤيتها من خلال المجهر.
يمكن أن تقيس اختبارات الدم أيضًا كمية الحديد في الدم، وتقييم الهيموغلوبين، واختبار الحمض النووي للطفل بحثًا عن جينات الهيموغلوبين غير الطبيعية، حيث إذا كان كلا الوالدين حاملين لاضطراب ثلاسيميا ألفا، يمكن للأطباء إجراء اختبارات على الجنين قبل الولادة، ويتم ذلك من خلال:
علاج الثلاسيميا
لا تحتاج الأشكال الخفيفة من الثلاسيميا إلى علاج، لكن بالنسبة للثلاسيميا المتوسطة إلى الشديدة، قد تشمل العلاجات ما يأتي:
1. عمليات نقل الدم
غالبًا تتطلب الأشكال الأكثر حدة من الثلاسيميا عمليات نقل دم متكررة ربما كل بضعة أسابيع، وبمرور الوقت يتسبب نقل الدم في تراكم الحديد في الدم مما قد يؤدي إلى تلف القلب والكبد والأعضاء الأخرى.
2. العلاج بالاستخلاب
هذا علاج لإزالة الحديد الزائد من الدم حيث قد يتراكم الحديد نتيجة لعمليات نقل الدم المنتظمة، ويمكن لبعض الأشخاص المصابين بالثلاسيميا الذين لا يخضعون لعمليات نقل الدم بانتظام أن يصابوا أيضًا بفرط الحديد.
للمساعدة في تخليص جسمك من الحديد الزائد قد تحتاج إلى تناول دواء عن طريق الفم، مثل: ديفيراسيروكس (Deferasirox)، أو ديفيريبرون (Deferiprone)، أو ديفيروكسامين (Deferoxamine) الذي يُعطى عن طريق الإبرة.
3. زرع الخلايا الجذعية
قد يكون زرع الخلايا الجذعية الذي يُسمى أيضًا بزرع نخاع العظم خيارًا في بعض الحالات، فعند الأطفال المصابين بالثلاسيميا الشديدة يمكن أن تلغي الحاجة إلى عمليات نقل الدم مدى الحياة والأدوية للسيطرة على الحديد الزائد.
يتضمن هذا الإجراء تلقي دفعات من الخلايا الجذعية من متبرع متوافق وعادةً يكون الأخ.
الوقاية من الثلاسيميا
في معظم الحالات لا يمكنك الوقاية من مرض الثلاسيميا حيث إذا كنت مصابًا بالثلاسيميا، أو إذا كنت تحمل جينًا من مرض الثلاسيميا فيجب التحدث مع مستشار وراثي للحصول على إرشادات إذا كنت ترغب في إنجاب الأطفال.
هناك شكل من أشكال التشخيص بمساعدة تقنية الإنجاب والذي يقوم بفحص الجنين في مراحله المبكرة بحثًا عن الطفرات الجينية المقترنة بالتخصيب في المختبر، قد يساعد هذا الآباء المصابين بالثلاسيميا أو حاملي جين الهيموغلوبين المعيب في إنجاب أطفال أصحاء.
يتضمن الإجراء استرجاع البويضات الناضجة وتخصيبها بالحيوانات المنوية في طبق في المختبر، ثم يتم اختبار الأجنة بحثًا عن الجينات المعيبة، ويتم فقط زرع الأجنة التي ليس لديها عيوب جينية في الرحم.
الأنواع الشائعة
يتكون الهيموغلوبين من نوعين من البروتينات: ألفا غلوبين، وبيتا غلوبين، حيث يحدث مرض الثلاسيميا عندما يكون هناك خلل في جين يساعد في التحكم في إنتاج أحد هذه البروتينات.
هناك نوعان رئيسيان من مرض الثلاسيميا:
1. ثلاسيميا ألفا
تحدث عندما يكون الجين أو الجينات المرتبطة ببروتين ألفا غلوبين مفقودة أو متغيرة، وتصيب ثلاسيميا ألفا في أغلب الأحيان الأشخاص من جنوب شرق آسيا، والشرق الأوسط، والصين، والمنحدرين من أصل أفريقي.
2. ثلاسيميا بيتا
عندما تؤثر عيوب جينية مماثلة على إنتاج بروتين بيتا غلوبين تحدث ثلاسيمسا بيتا، وتظهر غالبًا عند الصينيون، والآسيويون الآخرون، والأمريكيون الأفارقة.
هناك العديد من أشكال مرض الثلاسيميا وكل نوع له العديد من الأنواع الفرعية المختلفة، حيث يتضمن كل من ثلاسيميا ألفا وبيتا الشكلين الآتيين: الثلاسيميا الكبرى، والثلاسيميا الصغرى.
يجب أن ترث الخلل الجيني من كلا الوالدين للإصابة بمرض الثلاسيميا الكبرى، أما مرض الثلاسيميا الصغرى فيحدث إذا تلقيت الجين المعيب من أحد الوالدين فقط، فالأشخاص المصابون بهذا النوع من الاضطراب هم حاملو المرض وغالبًا لا تظهر عليهم أعراض.